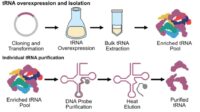
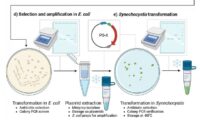
Protocols for SpudCell formation and experiments
Below is the list of protocols and procedures needed to assemble and analyze a SpudCell.
Read this first
Synthetic cell experiments are still more resource-intensive than growing natural cells. While our motivation for this research is to make biology a general purpose technology, usable freely by all, we are currently operating in the sandbox environment.
To grow a SpudCell, your lab needs to have core competencies in:
- liposome preparation
- PURE system expression
- aHL expression in liposomes
If you haven’t used any of those techniques before, we strongly suggest you successfully set up those first, before moving on to SpudCell experiments.
pREP Transcription–Translation and Phi29 Replication Reaction
- Purpose
This protocol describes the preparation and incubation of the pREP cell-free transcription–translation and DNA replication system used for protein expression, Phi29 genome replication, and related synthetic cell experiments.
- Materials
2.1 DNA preparation
- Plasmid DNA (mini-prepped, Epoch 2160250)
- PCR clean-up kit (Epoch 2360250)
- Lyophilizer for DNA concentration
2.2 Commercial reagents
- PURExpress Solution A (NEB E6800L)
- PURExpress Solution B (NEB E6800L)
- NxGen Phi29 DNA polymerase (Lucigen 30221-2)
- DTT (Goldbio DTT50)
- dNTPs (Denville Scientific CB4420-2)
- Amino acids (Sigma-Aldrich)
- Creatine phosphate (Sigma-Aldrich 27920-5G)
- Spermidine (Sigma-Aldrich S2526-5G)
- tRNA mix (E. coli, Sigma-Aldrich 10109550001)
- Stock Solutions (store at −80 °C)
3.1 10× Energy Mix
Prepare and aliquot:
- 700 mM potassium glutamate
- 79 mM magnesium glutamate
- 1 M HEPES pH 8.0
- 250 mM creatine phosphate
- 3.75 mM spermidine
- 5.18 g/L E. coli tRNAs
- 60 mM DTT
- 3.6 mM each L-amino acid (20 standard amino acids)
3.2 50× rNTP Mix
Prepare and aliquot:
- 18.75 mM ATP
- 12.5 mM GTP
- 6.25 mM CTP
- 6.25 mM UTP
- Reaction Setup
4.1 Final reaction composition (per 100 µL reaction)
Prepare reactions on ice.
|
Component
|
Final concentration
|
Volume (per 100 µL)
|
|
10× Energy Mix
|
1×
|
10 µL
|
|
50× rNTP Mix
|
1×
|
2 µL
|
|
PURExpress Solution A
|
0.1×
|
2 µL
|
|
PURExpress Solution B
|
60% v/v
|
60 µL
|
|
dATP
|
0.6 mM
|
variable (from stock)
|
|
dGTP
|
0.6 mM
|
variable
|
|
dCTP
|
0.6 mM
|
variable
|
|
dTTP
|
0.6 mM
|
variable
|
|
DTT
|
4 mM
|
variable
|
|
Phi29 DNA polymerase
|
0.4 U/µL
|
variable
|
|
Plasmid DNA
|
as required
|
variable
|
|
Nuclease-free water
|
—
|
to 100 µL
|
- Master Mix Preparation (recommended workflow)
5.1 Prepare bulk reaction master mix (excluding DNA and Phi29 polymerase if variable)
For N reactions, prepare:
- Combine in a sterile tube on ice:
- 10× Energy Mix (1× final)
- 50× rNTP Mix (1× final)
- PURExpress Solution A (0.1× final)
- PURExpress Solution B (60% final)
- dNTPs (0.6 mM each final)
- DTT (4 mM final)
- Nuclease-free water (to ~90% of final volume)
- Mix gently by pipetting (do not vortex).
- Keep on ice until use.
5.2 Final assembly per reaction
For each 100 µL reaction:
- Aliquot 90–95 µL of master mix into PCR tubes or plate wells.
- Add:
- Plasmid DNA (variable, typically 10–500 ng depending on experiment)
- Phi29 DNA polymerase to 0.4 U/µL final concentration
- Mix gently by pipetting.
- Briefly spin down (5–10 s).
- Incubation
- Incubate reactions at 30 °C.
- Standard reaction time: 8 hours.
- Avoid agitation unless specifically required for experimental variation.
- Storage and handling notes
- Keep all reagents on ice during setup.
- Avoid repeated freeze–thaw cycles of energy and nucleotide mixes.
- Prepare aliquots of master mixes when running multiple reactions.
- DNA should be highly purified and concentrated prior to use.
Plasmid Preparation, Cloning, and Usage for pREP / In Vitro Translation Experiments
- Purpose
This protocol describes plasmid sourcing, cloning strategies, purification workflow, and concentration handling for DNA templates used in pREP transcription–translation and related in vitro expression systems.
- Plasmid Sources
2.1 External plasmid sources (non-lab-cloned constructs)
- pLD1, pLD2, pLD3 plasmids (generous gift from Dr. Anthony Forster)
- α-hemolysin (αHL) and T7 RNA polymerase plasmids (generous gift from Dr. Vincent Noireaux)
2.2 Promoter system standardization
- All plasmids under T7Max promoter were cloned into a pre-existing lab vector system (previously described in ref. 67).
- This backbone contains:
- UTR1 sequence (optimized for bacterial in vitro translation)
- T500 transcription terminator (optimized for bacterial in vitro translation)
- Cloning Procedures
3.1 General cloning methods used
All cloning was performed using one of the following:
- Gibson Assembly
- NEB Gibson Assembly Master Mix (E2611)
- Site-directed mutagenesis
- Q5 High-Fidelity DNA Polymerase (NEB M0493)
- KLD Enzyme Mix (NEB M0554)
- Plasmid Amplification
4.1 Bacterial host strain
- DH5α E. coli cells used for plasmid amplification
4.2 Workflow
- Transform plasmid constructs into DH5α cells
- Grow under standard antibiotic selection conditions
- Isolate plasmid DNA from overnight cultures
- Plasmid Purification (Double-Clean Workflow)
5.1 Primary purification
- GenCatch™ Plasmid DNA Mini-Prep Kit (Epoch 2160250)
5.2 Secondary purification
- GenCatch™ PCR Cleanup Kit (Epoch 2360250)
5.3 Rationale
- Double purification was used based on empirical observations (colleagues’ reports) indicating improved in vitro translation yield with highly purified DNA templates.
- DNA Quantification and Normalization
6.1 Standard reaction concentrations
- Typical plasmid concentration in translation reactions:
- 10 nM final concentration per plasmid
6.2 Multi-plasmid conditions
- When multiple plasmids were used:
- Each plasmid was added at equal molar concentration
- Exception:
- Whole 90 kb genome experiments used 2.5 nM per plasmid
- DNA Handling for Multi-Plasmid Reactions
7.1 Problem addressed
- pREP reaction volume constraints result in limited free water volume after reagent addition.
7.2 Solution for high-plasmid load (>3 plasmids)
- Combine plasmid stock solutions into a single tube
- Lyophilize combined DNA mixture
- Resuspend concentrated DNA in minimal volume of nuclease-free water
- Typical resuspension volume: ~3 µL for a 30 µL final reaction
7.3 Outcome
- Enables high-complexity DNA inputs without increasing total reaction volume or diluting other components
- Standard Use in pREP Reactions
8.1 DNA input integration
- Add plasmid DNA to final reaction mix at desired molarity (typically 10 nM per construct unless specified otherwise)
8.2 Mixing strategy
- For multi-plasmid experiments:
- Pre-mix DNA stocks prior to reaction setup when necessary
- Ensure equal molar representation unless experimental design specifies otherwise
- Storage and Handling Notes
- Store plasmid stocks at −20 °C
- Avoid repeated freeze–thaw cycles
- Use double-purified DNA for optimal in vitro expression performance
- Maintain concentrated DNA stocks for multi-plasmid systems to minimize dilution effects
Synthetic Cell Liposome Preparation (Oil Extrusion Method)
- Purpose
This protocol describes the preparation, purification, and handling of synthetic cell liposomes used in cell-free synthetic biology experiments. Liposomes are prepared using a lipid thin-film method followed by oil hydration, emulsification, and differential centrifugation purification.
- Materials
2.1 Lipids
- DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; Avanti 850725P)
- DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine; Avanti 850375P)
- Cholesterol (Avanti 700100P)
2.2 Solutions
- Chloroform (anhydrous, for lipid stocks)
- Mineral oil
- Centrifuge buffer:
- 100 mM HEPES
- 200 mM glucose
- pH 8.0
- Wash buffer:
- 100 mM HEPES
- 250 mM glucose
- pH 8.0
- External reaction buffer (as required per experiment)
2.3 Equipment
- Glass vials (amber preferred)
- Glass syringes (for chloroform handling)
- Nitrogen gas supply
- Bath sonicator
- Vortexer with continuous shaking attachment
- Refrigerated centrifuge (4 °C capable)
- Gel loading pipette tips (preferred for oil removal)
- Lipid Composition
3.1 Standard formulation (“1× liposomes”)
- Total lipid concentration: 1 mM (excluding cholesterol from total lipid calculation)
- Molar ratio (DOPE:DOPC:Cholesterol): 1:3:1
3.2 Final composition per 1 mM lipid sample
- DOPE: 0.25 mM
- DOPC: 0.75 mM
- Cholesterol: 0.25 mM
- Preparation of Lipid Thin Film
4.1 Preparation of stock solutions
- Prepare lipid stocks in chloroform at 1 mg/mL
4.2 Thin film formation (typical 30 µL liposome batch)
- In an amber glass vial, combine:
- 5.58 µL DOPE stock
- 17.7 µL DOPC stock
- 2.9 µL cholesterol stock
- Mix using glass syringes only (avoid plastic contact with chloroform).
- Evaporate chloroform under gentle nitrogen stream for 6 hours to form a dry lipid film.
- Cap vials tightly and store at −20 °C until use.
- Liposome Formation (Oil Hydration and Emulsification)
5.1 Lipid hydration in oil phase
- Add dried lipid film to 200 µL mineral oil
- Incubate at 60 °C for 3 hours
- Vigorously shake during incubation
- Sonicate in bath sonicator for 10 minutes
- Incubate overnight at room temperature
- Continuous vigorous shaking (vortexer with attachment)
- Cool mineral oil suspension to 4 °C
- Maintain shaking during cooling
- Liposome Emulsification and Encapsulation
6.1 Emulsion formation
- Add 30 µL internal aqueous solution to mineral oil lipid suspension
- Vortex vigorously for 5 minutes at 4 °C
- Gradient Purification by Centrifugation
7.1 First centrifugation (phase separation)
- Carefully layer emulsion onto:
- 250 µL centrifuge buffer (100 mM HEPES, 200 mM glucose, pH 8)
- Centrifuge at:
- 18,000 × g (rcf), 4 °C, 15 minutes
- After spin:
- Carefully remove oil phase using gel loading tip
7.2 Liposome recovery and wash
- Collect liposomes from bottom aqueous phase using fresh pipette tip
- Transfer to:
- 250 µL pre-chilled wash buffer (100 mM HEPES, 250 mM glucose, pH 8)
- Centrifuge:
- 12,000 × g (rcf), 4 °C, 5 minutes
7.3 Final cleanup
- Remove any residual oil layer from top of wash buffer
- Transfer liposome pellet/phase to fresh tube
- Resuspend in final external reaction buffer
- Store or proceed immediately to experiments
- Temperature Control
- All steps from emulsification onward must be performed at 4 °C
- Maintain samples in cold room or on ice as appropriate
- Liposome Size Control (Extrusion Parameters)
Liposome size is defined by extrusion membrane pore size during initial lipid processing:
- 2 µm membrane → synthetic cell cycle experiments
- 0.8 µm membrane → DLS characterization samples
- 0.4 µm membrane → feeder liposomes
- Storage and Handling Notes
- Lipid stocks stored in chloroform must be kept in amber vials
- Thin lipid films stored at −20 °C
- Avoid repeated freeze–thaw of lipid films
- Use gel-loading tips for oil removal to maximize phase precision
- Prevent lipid oxidation by minimizing light exposure
Fluorescent Labeling of Synthetic Cell Liposomes
- Purpose
This protocol describes incorporation of fluorescent membrane dyes into lipid thin films for preparation of labeled synthetic cell liposomes used in imaging and tracking experiments.
- Materials
2.1 Lipid dyes (chloroform stocks)
All dyes are used as 100 µg/mL stocks in chloroform:
- NBD-PE
- (N-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine; Thermo Fisher N360)
- Rhodamine-PE (Lissamine™ Rhodamine B PE)
- (1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine; Thermo Fisher L1392)
- Cy5-PE
- (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Cyanine 5); Avanti 810335C)
- Cy7-PE
- (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Cyanine 7); Avanti 810337C)
- Labeling Strategy
3.1 Standard dye incorporation
- Dye concentration in liposomes:
- 0.1 mol% per dye relative to total lipid (excluding cholesterol from total lipid calculation)
3.2 Total dye loading rules
- Single dye system:
- Dual dye system:
- 0.1 mol% + 0.1 mol% = 0.2 mol% total dye
3.3 Effective concentration example
- For 1 mM liposomes:
- 0.1 mol% dye = 1 µM dye per lipid population
- Incorporation into Lipid Thin Film
4.1 General procedure
- During lipid thin film preparation (see liposome SOP section 4), add dye stock directly into chloroform lipid mixture.
- Mix dye stock with lipid chloroform solutions before solvent evaporation step.
- Proceed with nitrogen drying and thin film formation as standard.
- Dye Stock Addition (30 µL, 1 mM liposome batch)
For a standard 30 µL, 1 mM lipid preparation, add the following dye stock volumes directly into the chloroform lipid mixture:
|
Dye
|
Stock concentration
|
Volume per thin film
|
|
NBD-PE
|
100 µg/mL (chloroform)
|
2.86 µL
|
|
Rhodamine-PE
|
100 µg/mL (chloroform)
|
4.0 µL
|
|
Cy5-PE
|
100 µg/mL (chloroform)
|
3.79 µL
|
|
Cy7-PE
|
100 µg/mL (chloroform)
|
3.98 µL
|
- Thin Film Formation with Dye
- Combine lipid chloroform stocks as per standard liposome preparation protocol.
- Add appropriate dye stock volume (from Section 5) directly into lipid chloroform mixture.
- Mix thoroughly using glass syringes.
- Proceed with:
- Nitrogen drying (6 hours, gentle flow)
- Vacuum-free storage at −20 °C until use (sealed amber vial)
- Liposome Formation (Downstream Compatibility)
After dye incorporation, liposomes are prepared using the standard oil extrusion protocol:
- Dye-labeled lipid films are hydrated in mineral oil
- Follow identical emulsification, centrifugation, and purification steps as non-labeled liposomes
- Storage and Handling Notes
- Dye–lipid mixtures in chloroform must be protected from light (use amber vials)
- Avoid repeated freeze–thaw of lipid films
- Minimize oxygen exposure during drying to prevent dye degradation
- Store dried thin films at −20 °C in dark conditions
- All dye handling should be performed using glass syringes where possible
Preparation of Feeder Liposomes for Synthetic Cell Growth and Division Experiments
- Purpose
This protocol describes the preparation of Ni-NTA–functionalized feeder liposomes used in synthetic cell growth, division, and generation-tracking experiments. Feeder liposomes are designed to deliver biochemical components and enable membrane-mediated interactions such as fusion and cargo exchange.
- Materials
2.1 Lipids
- DOPE (Avanti 850725P)
- DOPC (Avanti 850375P)
- Cholesterol (Avanti 700100P)
- Ni-NTA lipid:
- 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] nickel salt
- Avanti 790404P
2.2 Lipid stock solutions
- Lipids prepared in chloroform (standard stocks)
- Ni-NTA lipid stock:
2.3 Equipment
- Avanti mini-extruder (Avanti 610023)
- Polycarbonate membranes (0.4 µm; Avanti 610007)
- Orbital shaker (4 °C compatible)
- Cold room (4 °C)
- Standard liposome preparation setup (see liposome SOP section 4)
- Lipid Composition
3.1 Base composition (all feeder liposomes)
- DOPE:DOPC:Cholesterol = 1:3:1 molar ratio
- Cholesterol is excluded from total lipid calculations
3.2 Functional lipid addition
- Ni-NTA lipid added at experiment-specific mol% relative to total lipid (excluding cholesterol)
3.3 Example calculation
- For 2 mM total lipid liposomes with 0.6 mol% Ni-NTA:
- Ni-NTA concentration = 12 µM
- Thin Film Preparation
4.1 Lipid mixture preparation
- Prepare lipid chloroform mixtures as per standard liposome thin film protocol.
- Add Ni-NTA lipid stock directly into chloroform lipid mixture prior to drying.
4.2 Example (30 µL, 2 mM feeder liposomes, 0.6 mol% Ni-NTA)
- Add 3.8 µL Ni-NTA chloroform stock (0.1 mg/mL) to lipid mixture.
4.3 Thin film formation
- Mix all lipid components in chloroform using glass syringes.
- Dry under gentle nitrogen flow to form thin film.
- Store dried film at −20 °C until use.
- Liposome Formation
5.1 Standard formation
- Prepare liposomes using the standard oil extrusion method (see liposome SOP section 4).
5.2 Hydration and emulsification
- Hydrate lipid film in mineral oil
- Proceed with heating, sonication, and shaking steps as in standard protocol
- Liposome Extrusion
6.1 Purpose
- Define feeder liposome size for controlled fusion and uptake behavior
6.2 Procedure
- Load liposome suspension into Avanti mini-extruder
- Extrude through 0.4 µm polycarbonate membranes
- Perform 9 extrusion passes
- Collect extruded liposomes under sterile cold conditions
- Post-Extrusion Equilibration
- Transfer liposomes to fresh tube
- Incubate at 4 °C for 10 minutes
- Gently shake on orbital shaker during equilibration
- Proceed immediately to experimental use
- Functional Loading for Cell Cycle Experiments
Each feeder liposome population may include:
- Ni-NTA lipid: 0.6 mol% (typical value unless otherwise stated)
- Generation counter oligo:
- 4 nM of one “bottom” oligonucleotide (see Methods section 12)
- Full pREP reaction mixture:
- Complete 1× pREP system (see Methods section 2)
- Standard Usage Conditions
- Typical reaction volume per condition:
- Lipid concentration:
- Used per generation in growth/division experiments unless otherwise specified
- Temperature and Handling
- All preparation and post-processing steps performed at 4 °C
- Maintain samples in cold room when possible
- Avoid prolonged warming after extrusion to preserve functionality
- Storage and Stability Notes
- Dried lipid films stored at −20 °C
- Ni-NTA lipid is sensitive to repeated freeze–thaw cycles
- Extruded liposomes should be used shortly after preparation for optimal performance
- Avoid extended storage after loading with biochemical components
Liposome Fusion Experiments
- Purpose
This protocol describes the preparation, tagging, mixing, incubation, and analysis of synthetic cell liposome fusion experiments using functionalized liposomes (e.g., protein tags, Ni-NTA systems, and αHL-mediated fusion).
- Materials
2.1 Liposomes
- Standard synthetic cell liposomes prepared via:
- Oil extrusion method (see liposome SOP section 4)
- Feeder liposome protocol (see feeder SOP section 6)
- Optional functionalized liposomes:
- Ni-NTA–functionalized liposomes
- Fluorescently labeled liposomes
2.2 Fusion tags and reagents
- Fusion tag systems (prepared according to published protocols75)
- α-hemolysin (αHL) system (expression-based fusion tag)
- Vamp-2 fusion proteins (see dedicated section in Methods)
2.3 Chemicals
- Triton X-100 (Acros Organics AC215682500)
2.4 Equipment
- Orbital shaker (room temperature and 30 °C capable incubator setup)
- PCR tubes
- 96-well plates (for endpoint fluorescence readout)
- Fluorescence plate reader
- Incubator set to 30 °C
- Liposome Preparation (Pre-Fusion)
3.1 Base preparation
- Liposomes prepared as described in:
- Standard liposome SOP (Methods section 4)
- Feeder liposome SOP (Methods section 6)
3.2 Standard concentration
- All fusion samples prepared at:
- 1 mM total lipid concentration
- Exceptions:
- Only as specified in experimental variations
- Fusion Tag Functionalization
4.1 External fusion tag decoration
- Prepare liposomes as described above.
- Incubate liposomes with fusion tags (per protocol75).
- Incubation conditions:
- 30 minutes at room temperature
- Gentle tumbling on orbital shaker
4.2 αHL-based fusion system
- Prepare synthetic cells expressing αHL as described in expression protocols.
- Incubation conditions:
- 3 hours at 30 °C
- Gentle shaking on orbital shaker inside incubator
- Sample Mixing for Fusion Assays
5.1 Standard mixing procedure
- After tag incubation or αHL expression:
- Mix liposome populations in equal volumes, unless otherwise specified
- Each liposome population is typically:
5.2 Mixing format
- Perform mixing in:
- Use gentle pipetting to avoid mechanical rupture
- Fusion Incubation
6.1 Standard conditions
- Incubate mixed samples:
- 30 °C
- Gentle shaking on orbital shaker inside incubator
- Standard incubation time:
- 6–12 hours (depending on experiment)
- Typical default: 12 hours
- Post-Fusion Analysis
7.1 Fluorescence measurement
- Transfer samples to 96-well plates
- Measure fluorescence using plate reader
7.2 Molecular analysis (if required)
- For RT-PCR, qPCR, or Western blot:
- Lyse liposomes using:
- Proceed with downstream molecular assays
- Standard Reaction Conditions Summary
- Lipid concentration:
- 1 mM total lipid per sample
- Includes Ni-NTA and/or dyes when applicable
- Mixing ratio:
- Typically 1:1 volume ratio
- Incubation temperature:
- Shaking:
- Gentle orbital shaking throughout incubation
- Storage and Handling Notes
- Prepare fusion samples fresh when possible
- Maintain gentle handling to prevent premature rupture
- Avoid vigorous vortexing after fusion tag binding
- Keep samples at 30 °C only during incubation phases
FRET-Based Membrane Growth and Lipid Mixing Assay
- Purpose
This protocol describes fluorescence resonance energy transfer (FRET)–based quantification of membrane growth and lipid mixing in synthetic cell liposomes. The assay monitors dilution of membrane dyes upon liposome fusion and membrane expansion.
- Principle
Membrane growth is quantified by measuring dilution of FRET pairs embedded in lipid bilayers. Upon fusion with unlabeled membranes, FRET efficiency decreases, leading to increased donor fluorescence.
- Dye dilution → decreased FRET → increased donor signal
- Signal is calibrated to % lipid mixing using defined standards
- Materials
3.1 Lipid dyes (FRET pairs)
- NBD–Rhodamine pair:
- NBD-PE (donor)
- Lissamine Rhodamine-PE (acceptor)
- Cy5–Cy7 pair:
- Cy5-PE (donor)
- Cy7-PE (acceptor)
3.2 Liposomes
- Prepared via standard liposome thin film and extrusion methods:
- Liposome SOP (section 4)
- Fluorescent labeling SOP (section 5)
3.3 Equipment
- Avanti mini-extruder (Avanti 610023)
- 0.4 µm polycarbonate membranes (Avanti 610007)
- Fluorescence plate reader or spectrofluorometer
- Standard incubation equipment
- FRET Pair Selection Rules
4.1 Pair usage
- NBD–Rhodamine
- Used when no GFP signal is present
- Cy5–Cy7
- Used when GFP is present (avoids spectral overlap with NBD)
- Experimental Design
5.1 Liposome populations
- Population A:
- Labeled with both donor and acceptor FRET dyes
- Population B:
- Unlabeled (no membrane dyes)
5.2 Mixing principle
- Fusion or membrane growth leads to:
- Dilution of FRET dyes
- Decreased FRET efficiency
- Increased donor fluorescence
- FRET Liposome Preparation
- Prepare lipid thin films (see liposome SOP section 4)
- Incorporate FRET dyes during chloroform stage (see dye labeling SOP section 5)
- Form liposomes using standard oil extrusion method
- Extrude through:
- 0.4 µm polycarbonate membranes
- Using Avanti mini-extruder
- Perform standard extrusion procedure
- FRET Calibration Standards
7.1 Purpose
- Convert fluorescence signal into % lipid mixing
7.2 Calibration series
Prepare control liposomes at defined dye dilution levels:
- 0% membrane mixing
- 1 mM liposomes
- 0.1 mol% donor + 0.1 mol% acceptor (total 0.2 mol% FRET dyes)
- 50% membrane mixing
- 1 mM liposomes
- 0.05 mol% donor + 0.05 mol% acceptor
- Additional calibration points prepared analogously by proportional dilution of both dyes
7.3 Preparation method
- All calibration samples:
- Prepared from thin lipid films
- Extruded through 0.4 µm membranes (as above)
- FRET Measurement
8.1 Signal acquisition
- Measure fluorescence using donor-specific wavelengths:
- NBD or Cy5 emission channels depending on system used
8.2 Spectral configuration
- Refer to Table S5 for excitation/emission settings
- Data Interpretation
9.1 Signal readout
- Increased donor fluorescence indicates:
- Reduced FRET efficiency
- Increased membrane mixing or growth
9.2 Calibration
- Raw fluorescence values are converted to:
- % lipid mixing using calibration curve (Figure S76)
- Storage and Handling Notes
- Protect dye-labeled liposomes from light
- Prepare fresh calibration standards when possible
- Avoid photobleaching during incubation and measurement
- Maintain consistent extrusion conditions across samples
Synthetic Cell Cycle with Feeder Liposome Growth and Mechanical Division
- Purpose
This protocol describes a multi-generation synthetic cell cycle system combining feeder-liposome–driven growth, pREP-based cell-free expression, and mechanical division via extrusion. The system supports tracking over 5 sequential generations with molecular and fluorescence-based readouts.
- Materials
2.1 Liposomes
- Synthetic cells prepared via standard liposome protocol (Methods section 4)
- Feeder liposomes prepared via feeder protocol (Methods section 6)
- Ni-NTA–functionalized feeder liposomes
2.2 Reaction components
- Complete pREP transcription–translation system (Methods section 2)
- Plasmid DNA templates (Methods section 3)
- T4 DNA ligase (for generation counter assembly)
- Oligonucleotides for generation counter (Methods section 12)
2.3 Reagents for analysis
- Triton X-100 (0.1% final for lysis; Acros Organics AC215682500)
- EDTA (0.5 M stock, used to 10 mM final)
- DpnI restriction enzyme (NEB R0176)
- rCutSmart buffer (NEB B6004)
2.4 Equipment
- Avanti mini-extruder (Avanti 610023)
- 2 µm membranes (Whatman 76457-184)
- Orbital shaker (30 °C incubator compatible)
- Plate reader (Molecular Devices SpectraMax)
- Centrifuge and lyophilizer
- Heat block / incubator (65–80 °C)
- Initial Synthetic Cell Preparation (Generation 0)
3.1 Liposome formation
- Prepare synthetic cells as described in liposome SOP (section 4)
3.2 Initial reaction composition (per 30 µL)
- Complete 1× pREP reaction mix (Methods section 2)
- 4 µL plasmid DNA mix (Methods section 3; typically 10 nM each)
- 1 µL “top strand” generation counter oligos (Methods section 12)
- 1 µL T4 DNA ligase
3.3 Loading conditions
- Total internal volume: 30 µL
- DNA templates present from start of experiment
- Generation 0 Incubation
- Incubate samples:
- 30 °C
- 12 hours
- Gentle tumbling on orbital shaker
- This defines:
- Feeder Liposome Addition (Generation Progression)
5.1 First growth step (Generation 1 initiation)
- Mix:
- 30 µL generation 0 synthetic cells
- 30 µL feeder liposomes (2 mM total lipid)
- Incubate:
- 30 °C
- 12 hours
- Gentle shaking
- Feeder Liposome Composition
Each feeder liposome batch contains:
- 2 mM total lipid concentration
- 4 nM “bottom strand” generation counter oligo (Methods section 12)
- Complete 1× pREP reaction mixture
- No DNA template included
- Ni-NTA–functionalized membrane lipids (Methods section 6)
- Mechanical Division (Extrusion-Based Splitting)
7.1 End of each generation cycle
- After 12-hour incubation:
- Extrude liposome mixture through:
- 2 µm polycarbonate membrane
- Avanti mini-extruder (610023)
- This step defines completion of each generation
- Iterative Growth Cycles (Generations 1–5)
For each subsequent generation:
- Mix extruded population (30 µL) with:
- 30 µL fresh 2 mM feeder liposomes
- Incubate:
- 30 °C
- 12 hours
- Gentle shaking
- Repeat:
- Extrusion (2 µm membrane)
- Feeding step
- Incubation
- Analytical Workflow (Starting Generation 3)
After each cycle (post-incubation + extrusion):
9.1 Fluorescence measurement
- Measure GFP fluorescence:
- Plate reader (Molecular Devices SpectraMax)
9.2 Liposome lysis for molecular assays
- Lyse samples with:
- 0.1% Triton X-100 (final)
- Add EDTA:
- Final concentration: 10 mM
- Heat inactivate:
9.3 DNA digestion (new DNA quantification)
- Treat with DpnI:
- 5 units per reaction
- rCutSmart buffer (1×)
- Incubate:
- Heat inactivate:
9.4 Sample concentration and preparation
- Lyophilize samples
- Resuspend in:
- 15 µL nuclease-free water
- Use for downstream analysis:
- DNA abundance
- mRNA levels
- generation counter RNA
- Molecular Readouts
- DNA quantification
- mRNA abundance
- generation counter system output
- Control Experiments
11.1 Fluorescent protein control
- Experiments performed with:
- GFP-expressing synthetic cells
- GFP + mCherry competition experiments
11.2 No-GFP control condition
- Full 5-generation cycle repeated without GFP expression
11.3 Outcome comparison
- DNA and RNA abundance compared between conditions
- Membrane dynamics validated as unchanged
- Incubation Conditions Summary
- Temperature: 30 °C
- Duration per generation: 12 hours
- Mixing: gentle orbital shaking
- Division: mechanical extrusion (2 µm membrane)
Generation Counter System (Oligo Ligation and Readout)
- Purpose
This protocol describes the assembly, optimization, and quantification of a synthetic “generation counter” system based on ligation of RNA oligonucleotides with complementary sticky ends. The system is used to track synthetic cell generations in liposome-based cell cycle experiments.
- Materials
2.1 Oligonucleotides
- Generation counter oligos (Table S3)
- Custom synthesized RNA oligos (IDT DNA)
- Used without additional purification
2.2 Enzymes
- T4 DNA ligase (NEB M0202)
2.3 Nucleotides
- dATP, dGTP, dCTP, dTTP (Denville Scientific CB4420-2)
2.4 Buffer components (pREP-compatible ligation conditions)
- Potassium glutamate
- Magnesium glutamate
- HEPES (pH 8.0)
- Creatine phosphate
- Spermidine
- E. coli tRNAs
- DTT
- ATP, GTP, CTP, UTP (Larova nucleotides)
2.5 Equipment
- Thermal incubator (30 °C)
- Lyophilizer
- RT-qPCR system (Methods section 15)
- Standard molecular biology pipettes and tubes
- Oligonucleotide Preparation
3.1 Stock preparation
- Each generation counter oligo stored at:
- 20 nM in nuclease-free water
3.2 Working mixture preparation
- Mix 1 µL of each 20 nM oligo stock
- Combine into a single oligo pool
- Lyophilize mixture
- Resuspend in:
- Use as master generation counter input
- Reaction Setup (20 µL total volume)
4.1 Standard reaction composition
Each 20 µL ligation reaction contains:
- 1 µL T4 DNA ligase (NEB M0202)
- Generation counter oligos:
- 1 nM each oligo (from pooled mixture)
- dNTPs:
- 0.6 mM each of dATP, dGTP, dCTP, dTTP
- pREP-compatible energy mix (1× final), containing:
- 70 mM potassium glutamate
- 7.9 mM magnesium glutamate
- 0.1 M HEPES pH 8.0
- 25 mM creatine phosphate
- 0.375 mM spermidine
- 0.5 g/L E. coli tRNAs
- 10 mM DTT
- 0.3775 mM ATP
- 0.25 mM GTP
- 0.125 mM CTP
- 0.125 mM UTP
- Reaction Design Notes
- Buffer conditions are adapted from T4 DNA ligase standard buffer (NEB B0202S), but modified to match pREP compatibility.
- Non-essential transcription–translation components are included to ensure compatibility with downstream synthetic cell conditions.
- Reaction conditions prioritize in vivo–relevant mimicry over enzymatic optimality.
- Incubation Conditions
- Incubate ligation reactions at:
- Note:
- This temperature is suboptimal for ligation efficiency
- It is intentionally used to match synthetic cell cycle conditions
- Post-Reaction Analysis
7.1 RT-qPCR quantification
- Use ligation reaction as template:
- 4.5 µL per RT-qPCR reaction (from 20 µL total)
- Perform RT-qPCR according to Methods section 15
7.2 Output measured
- Full-length generation counter ligation products
- Relative abundance across conditions
- Integration into Liposome Cell Cycle System
8.1 Synthetic cell (top strand loading)
- Each starting “parent” liposome contains:
- 1 nM of each “top strand” oligo
- Preparation:
- Mix oligos from 20 nM stocks
- Lyophilize pooled mixture
- Resuspend in 1 µL
- Add to liposome lumen during assembly (Methods section 4)
8.2 Feeder liposome loading (bottom strand delivery)
- Each feeder liposome contains:
- 4 nM of one “bottom strand” oligo per generation
- Rationale:
- Higher concentration compensates for dilution effects during fusion with larger synthetic cells
- Experimental Use in Cell Cycle Assays
- Generation counter system is integrated into:
- Liposome growth and division experiments (Methods section 11)
- Tracking:
- Full-length counter product measured after:
- Generation 3
- Generation 4
- Generation 5
- Storage and Handling Notes
- RNA oligos stored at −20 °C (20 nM stocks)
- Avoid repeated freeze–thaw cycles
- Lyophilized oligo mixtures should be freshly prepared when possible
- Maintain RNase-free conditions throughout handling
Genetically Encoded Growth and Division (FLAG-tag–Mediated System)
- Purpose
This protocol describes a genetically encoded synthetic cell growth and division system using FLAG-tagged αHL, antibody–streptavidin linker chemistry, azide-functionalized membrane immobilization, and feeder liposome–driven growth.
- Materials
2.1 Synthetic cells and liposomes
- Synthetic cells prepared via standard liposome protocol (Methods section 4)
- Feeder liposomes prepared via feeder protocol (Methods section 6)
- Ni-NTA–functionalized feeder liposomes
- pREP reaction components included in feeder system
2.2 Genetic and protein components
- Complete synthetic genome (as specified in experiment)
- FLAG-tagged α-hemolysin (αHL), 10 nM final concentration
2.3 Immobilization chemistry
- Azido lipids (1 mol% in synthetic cell membrane; Methods section 20)
- Streptavidin (5 µM stock used in reactions)
- Biotin–FLAG antibody linker (Abcam ab173832)
2.4 Buffers and reagents
- 100 mM HEPES, pH 8.0
- Triton X-100 (0.1% final; Acros Organics AC215682500)
2.5 Solid support
- Magnetic beads (5 mg per reaction)
2.6 Equipment
- Magnetic PCR plate or magnetic rack
- Incubator with orbital shaker (30 °C)
- PCR tubes
- Lyophilizer
- qPCR system (Methods section 14)
- Reagent Preparation
3.1 Biotin–FLAG antibody stock
- Manufacturer stock: 1 mg/mL (~28 µM, assuming 35 kDa)
- Diluted working stock:
- Stored at:
3.2 Streptavidin working stock
- Used at:
- Final concentration in reaction:
- Pre-Assembly of Linker Complex
- Mix:
- Streptavidin (5 µM stock)
- Biotin–FLAG antibody linker (20 µM stock)
- Incubate:
- 30 °C for 10 minutes
- Gentle mixing
- This forms streptavidin–antibody linker complex for immobilization
- Synthetic Cell Preparation (Parent Population)
5.1 Liposome composition
- Prepared as described in liposome SOP (section 4)
- Includes:
- Complete synthetic genome
- 10 nM FLAG-tagged αHL
- 1 mol% azido lipid (for immobilization)
5.2 Immobilization setup
- Synthetic cells are immobilized onto magnetic beads prior to feeding step
- Feeder Liposome Preparation
- Prepared as described in feeder liposome SOP (section 6)
- Contains:
- Ni-NTA lipid functionalization
- Complete pREP reaction mixture
- Immobilization and Feeding Reaction Setup
- Add to each reaction:
- 5 mg magnetic beads with immobilized synthetic cells
- 30 µL feeder liposomes
- 1.2 µL pre-incubated streptavidin–biotin–FLAG linker solution
- Generation counter configuration:
- Immobilized “parent” cells contain:
- gen1, gen2, gen3 “top strand” oligos
- Feeder liposomes contain:
- gen1, gen2, gen3 “bottom strand” oligos
- Detection strategy:
- Full-length product detected using gen3 primers only
- Multiple oligos used to generate sufficient amplicon length for detection robustness
- Incubation Conditions
- Temperature: 30 °C
- Duration: 12 hours
- Mixing:
- Gentle shaking in PCR tubes on orbital shaker inside incubator
- Magnetic Separation Workflow
9.1 Post-incubation separation
- Collect beads using magnetic PCR plate
- Remove supernatant carefully
- Wash beads with:
- 30 µL of 100 mM HEPES (pH 8)
- Combine:
- Wash solution + original supernatant fraction
- Fraction Processing
10.1 “Free (off-beads)” fraction
- Liposomes in supernatant + wash combined
- Lyse:
- 0.1% Triton X-100
- 10 min incubation
- Lyophilize to concentrate
- Resuspend in:
10.2 “Bead-attached” fraction
- Resuspend beads in:
- 50 µL 100 mM HEPES (pH 8)
- Lyse attached liposomes:
- 0.1% Triton X-100
- 10 min incubation
- Remove lysate from beads
- Lyophilize
- Resuspend in:
- Molecular Analysis
- Analyze both fractions using qPCR:
- Outputs:
- Relative abundance of generation counter products
- Distribution between immobilized vs released populations
- Storage and Handling Notes
- Maintain samples at 4 °C before incubation
- Avoid bead drying prior to lysis steps
- Handle azido-functionalized liposomes carefully to preserve surface chemistry
- Perform all post-lysis concentration steps promptly
Western Blot Analysis of Protein Expression
- Purpose
This protocol describes SDS-PAGE and Western blot analysis of proteins expressed in solution or within synthetic cell liposomes, including sample preparation, electrophoresis, transfer, immunodetection, and chemiluminescent imaging.
- Materials
2.1 Samples
- Cell-free expression reactions in solution
- Synthetic cell liposome samples (Methods section 4 and related protocols)
2.2 Lysis reagent (for liposome samples)
- Triton X-100 (0.1% final concentration)
2.3 SDS-PAGE reagents
- 2× SDS loading buffer:
- 100 mM Tris-HCl
- 2.5% SDS
- 20% glycerol
- 4% β-mercaptoethanol
- 0.1% bromophenol blue
- Acrylamide:Bis-acrylamide gel (37.5:1)
- SDS running buffer:
- 25 mM Tris
- 192 mM glycine
- 3.5 mM SDS
2.4 Transfer system
- Nitrocellulose membrane (0.2 µm)
- Transfer buffer:
- 25 mM Tris
- 192 mM glycine
2.5 Immunoblotting reagents
- TBST buffer:
- 20 mM Tris (pH 7.4)
- 150 mM NaCl
- 0.05% Tween-20
- Nonfat dry milk (blocking reagent, 5%)
- Mouse IgG1 anti-His primary antibody (BioLegend 652505)
- HRP-conjugated goat anti-mouse IgG1 secondary antibody (BioLegend 405306)
2.6 Detection system
- SuperSignal chemiluminescent substrate (Thermo Scientific 34577)
2.7 Equipment
- Mini-PROTEAN electrophoresis system (Bio-Rad)
- PowerPac 3000 power supply (Bio-Rad)
- Thermocycler or heat block (95 °C capability)
- Horizontal rocker
- ChemiDoc MP imaging system (Bio-Rad)
- Image Lab software v5.2.1
- Sample Preparation
3.1 Solution samples (no vesicles)
- Mix directly with loading buffer (1:1 ratio with 2× SDS loading buffer)
3.2 Liposome samples
- Lyse samples with:
- Proceed to mixing with loading buffer (1:1 with 2× SDS loading buffer)
- Denaturation
- Incubate samples in loading buffer at:
- 95 °C for 5 minutes
- Use thermocycler or heat block
- SDS-PAGE Electrophoresis
- Load samples onto:
- 37.5:1 acrylamide:bis-acrylamide gels
- Run gel using Mini-PROTEAN system
- Conditions:
- 100 V for 60 minutes
- In 800 mL SDS running buffer
- Protein Transfer
- Transfer proteins to:
- 0.2 µm nitrocellulose membrane
- Conditions:
- 100 V for 60 minutes
- In 1 L transfer buffer
- Membrane Blocking
- Incubate membrane in:
- Conditions:
- 60 minutes
- Horizontal rocking
- Primary Antibody Incubation
- Prepare antibody solution:
- Mouse IgG1 anti-His antibody (BioLegend 652505)
- Dilution: 1:5000 in 5% milk/TBST
- Incubate membrane:
- 60 minutes
- Horizontal rocker
- Washing Steps
- Wash membrane:
- 3× quick rinses with TBST
- 3× 10-minute washes with TBST
- Secondary Antibody Incubation
- Prepare solution:
- HRP-conjugated goat anti-mouse IgG1 (BioLegend 405306)
- Dilution: 1:5000 in 5% milk/TBST
- Incubate membrane:
- 60 minutes
- Horizontal rocking
- Final Washes
- Repeat washing:
- 3× TBST rinses
- 3× 10-minute TBST washes
- Chemiluminescent Detection
- Apply:
- SuperSignal substrate (Thermo Scientific 34577)
- Develop blot according to manufacturer instructions
- Imaging
- Image membrane using:
- ChemiDoc MP Imaging System (Bio-Rad)
- Software:
- Storage and Handling Notes
- Keep membranes hydrated during all antibody steps
- Avoid drying of nitrocellulose membrane before detection
- Prepare antibody dilutions fresh for optimal signal quality
- Ensure complete removal of Triton X-100 prior to electrophoresis
Size Exclusion Chromatography (SEC) Purification of Liposomes
- Purpose
This protocol describes gravity-driven size exclusion chromatography (SEC) used to separate liposomes from unencapsulated solutes following synthetic cell or liposome preparation.
- Materials
2.1 Chromatography column
- Poly-Prep Chromatography Column (Bio-Rad 7311550; 10 mL plastic column)
2.2 Size exclusion resin
- Sepharose 4B beads (Sigma-Aldrich 4B200-1L)
- Bead size: 45–165 µm
- Stored as ethanol slurry
2.3 Buffers
2.4 Equipment
- Fraction collector (Gilson FC203B)
- 96-well collection plates
- Gravity flow setup
- Column Packing
- Fill 10 mL chromatography column with Sepharose 4B ethanol slurry.
- Allow beads to settle by gravity.
- Drain ethanol solvent completely.
- Column Equilibration
- Wash column with ≥4 column volumes of:
- Avoid disturbing the bead bed during washing.
- After final wash:
- Drain buffer until liquid level is just above bead surface.
- Sample Loading
- Prepare liposome sample (typically 20–100 µL).
- Gently apply sample dropwise across the entire column surface.
- Minimum effective loading volume:
- Prevent disruption of bead surface during loading.
- Elution
- Immediately add 50 mM HEPES buffer to cover bead surface after loading.
- Ensure column does not dry out at any point.
- Allow elution to proceed by gravity flow only.
- Do not apply pressure or accelerate flow.
- Fraction Collection
- Collect eluate into 96-well plates using:
- Gilson FC203B fraction collector
- Collect at least:
- 48 fractions per column run
- Ensure full separation of:
- Liposome-containing fractions
- Free (unencapsulated) solute fractions
- Post-Purification Processing
- Analyze fractions using plate reader (initial screening).
- Identify liposome-containing fractions based on signal profiles.
- If required:
- Recover selected fractions from wells
- Concentrate via lyophilization
- Perform downstream biochemical or fluorescence assays
- Critical Handling Notes
- Do not disturb bead bed at any stage after equilibration
- Maintain continuous buffer coverage to prevent column drying
- Apply samples gently to avoid mixing with resin surface
- Use gravity flow exclusively for reproducible separation
Single-Cell Plasmid and Generation Counter Product Abundance Analysis
- Purpose
This protocol describes isolation, dilution, and molecular screening of single synthetic cells to quantify plasmid distribution and generation counter products after growth and division cycles.
- Materials
2.1 Synthetic cells
- Synthetic cell populations after growth/division experiments
- Feeder liposome removal system (dialysis; Methods section 24)
2.2 Molecular biology reagents
- 16S rRNA primers (Table S2)
- dNTPs (Denville Scientific CB4420-2)
- Superscript IV Reverse Transcriptase (Invitrogen 18090010)
- Superscript IV RT buffer (5×)
- RNase Inhibitor Murine (NEB M0314S)
- OneTaq 2× Master Mix (NEB M0482)
- Chai Green Dye (Chai R01200S)
2.3 Equipment
- Cold room (4 °C)
- Thermocycler / heat blocks (52–80 °C capability)
- qPCR systems:
- Chai Open qPCR machine (single or dual channel)
- CFX Opus 384 System
- −80 °C storage
- 384-well PCR workflow setup
- Sample Preparation
3.1 Removal of feeder liposomes
- Dialyze synthetic cell samples to remove feeder liposomes (see Methods section 24).
3.2 Normalization
- Adjust sample concentration to:
- OD ~0.2 equivalent synthetic cell concentration
3.3 Single-cell dilution strategy
- Perform sequential dilution at 4 °C (cold room).
- Work rapidly to minimize leakage near critical aggregation concentration (CAC).
- Target dilution:
- ~1 synthetic cell per 1 µL
- Collect:
- 0.8 µL per aliquot
- Total: 384 aliquots
- Single-Cell Occupancy Verification
4.1 Rationale
- Many aliquots are expected to be empty
- Presence of ribosomes (16S rRNA) used as proxy for cell presence
4.2 Screening marker
- E. coli 16S rRNA amplification used to confirm cell-containing wells
- Reverse Transcription (RT) Setup (10 µL reaction)
For each putative single-cell aliquot:
- Prepare initial mix:
- 0.8 µL sample
- 0.5 µL 2 µM forward 16S primer
- 0.5 µL 10 mM dNTPs
- 4.7 µL nuclease-free water
- Incubation:
- 65 °C for 1 minute
- Immediately place on ice for ≥5 minutes
- Add RT components:
- 2 µL 5× Superscript IV buffer
- 0.5 µL 100 mM DTT
- 0.5 µL Superscript IV RT (200 U/µL)
- 0.5 µL RNase inhibitor (40 U/µL)
- RT incubation:
- Enzyme inactivation:
- Storage:
- Retain remaining 8 µL at −80 °C
- PCR Amplification (OneTaq)
- Prepare 20 µL PCR reaction:
- 2 µL RT product (template)
- OneTaq 2× Master Mix (NEB M0482)
- Chai Green Dye (1× final concentration)
- Run PCR following manufacturer-recommended cycling conditions.
- qPCR and Detection
- Analyze amplification using:
- Chai Open qPCR system (single or dual channel), or
- CFX Opus 384 System
- Determine:
- Presence/absence of synthetic cell per aliquot (via 16S signal)
- Positive wells selected for downstream plasmid and generation counter analysis
- Downstream Interpretation
8.1 Cell-containing selection
- Only 16S-positive wells are considered valid single-cell samples
8.2 Quantification targets
- Plasmid copy distribution
- Generation counter product abundance
- Critical Handling Notes
- Maintain all dilution steps at 4 °C
- Minimize handling time during serial dilution
- Avoid membrane leakage by rapid processing near CAC conditions
- Expect high fraction of empty wells in 384-well sampling scheme
- Store intermediate RT products at −80 °C when needed
Genetically Encoded Division of Synthetic Cells (Streptavidin–Biotin NTA System)
- Purpose
This protocol describes a genetically encoded division system for synthetic cells using streptavidin–biotin–NTA crosslinking to induce membrane organization and division-like behavior. The system supports both free-solution and immobilized bead-based formats, including competitive growth experiments.
- Materials
2.1 Proteins and linkers
- Streptavidin (BioLegend 280302; 1.0 mg/mL ~19 µM stock)
- Biotin-NTA linker (VWR 90074)
2.2 Buffers
- 50 mM HEPES, pH 8.0
- 100 mM HEPES, pH 8.0
- PBS
2.3 Liposomes
- Synthetic cells prepared as in cell cycle SOP (Methods section 11)
- Feeder liposomes (when applicable, excluded from division system)
- Liposomes without generation counter oligos (for division-only experiments)
2.4 Solid support
- Magnetic beads (5 mg per sample)
- Azide-blocked immobilization surface (when used)
2.5 Molecular biology reagents
- Triton X-100 (0.1%; Acros Organics AC215682500)
- qPCR reagents (Methods section 14)
2.6 DNA markers (competitive experiments only)
- CMV-GFP plasmid (10 nM encapsulated)
- CMV-mCherry plasmid (10 nM encapsulated)
2.7 Equipment
- Orbital shaker (30 °C incubator compatible)
- PCR tubes
- Magnetic PCR rack/plate
- DLS instrument (Methods section 19)
- qPCR system
- Reagent Preparation
3.1 Streptavidin working stock
- Start from:
- Dilute in:
- Final working concentration:
- Store at:
3.2 Biotin-NTA linker working stock
- Resuspend in:
- Final concentration:
- Store at:
- Stoichiometric Design (Binding Optimization)
- Liposome concentration reference:
- ~6.71 × 10¹⁰ liposomes per mL at 1 nM (1 µm diameter)
- ≈ 0.003 pmol liposomes per 30 µL sample
- Target loading:
- ~1000 streptavidin molecules per liposome
- Final working concentrations:
- Streptavidin: 0.1 µM (3 pmol per 30 µL)
- Biotin-NTA linker: 0.4 µM (4× excess relative to streptavidin binding sites)
- Pre-Complex Formation
- Mix per sample:
- 0.6 µL of 5 µM streptavidin stock
- 0.6 µL of 20 µM biotin-NTA linker stock
- Incubation:
- 30 °C for 10 minutes
- Gentle mixing
- Synthetic Cell Preparation
- Liposomes prepared as in cell cycle SOP (Methods section 11)
- For division-only experiments:
- No generation counter oligos included
PART A — Free-Solution Division Experiments
7A. Reaction Setup
- Combine:
- 30 µL synthetic cells (1 mM total lipid)
- 1.2 µL pre-incubated streptavidin–biotin-NTA complex
8A. Incubation
- Conditions:
- 30 °C
- 12 hours
- Gentle shaking in PCR tubes
9A. Post-Incubation Processing
- Dilute samples to:
- 0.1 mM total lipid in PBS
- Analyze by:
- Dynamic light scattering (DLS; Methods section 19)
PART B — Immobilized Division Experiments
7B. Bead Immobilization Setup
- Combine:
- 5 mg magnetic beads with immobilized 30 µL synthetic cells (azide-blocked)
- 1.2 µL streptavidin–biotin-NTA pre-complex
8B. Incubation
- 30 °C for 12 hours
- Gentle shaking in PCR tubes
9B. Fraction Separation
- Magnetically separate beads
- Collect supernatant (“daughter fraction”)
- Wash beads with:
- 50 µL 100 mM HEPES pH 8.0
- Combine:
- Wash + original supernatant → daughter fraction
10B. Fraction Processing
10B.1 Daughter fraction
- Lyse with:
- 0.1% Triton X-100 (10 min)
- Lyophilize
- Resuspend in:
10B.2 Bead-bound fraction
- Resuspend beads in:
- 100 µL 100 mM HEPES pH 8.0
- Lyse:
- 0.1% Triton X-100 (10 min)
- Remove lysate
- Lyophilize
- Resuspend in:
- Analysis
- Quantify both fractions using qPCR (Methods section 14)
- Outputs:
- Distribution of synthetic cell material between daughter and retained populations
PART C — Competitive Growth Experiments
12C. System Setup
- Prepare two bead-immobilized populations:
- T7Max αHL synthetic cells
- T7 αHL synthetic cells
- Each population:
- 5 mg beads + 30 µL synthetic cells
13C. Genetic Markers
To distinguish populations:
- T7 cells:
- T7Max cells:
- 10 nM CMV-mCherry plasmid
Note:
- CMV promoter ensures no bacterial expression burden
- Used solely as neutral lineage markers
14C. Competition Reaction
- Mix both populations
- Add streptavidin–biotin-NTA complex as above
- Incubate:
- 30 °C for 12 hours
- Gentle shaking
15C. Post-Reaction Processing
- Same bead separation and fraction workflow as Section 9B
- Wash and resuspension volumes doubled relative to single-population experiments
- Analyze daughter fraction via qPCR and marker plasmid readout
- Storage and Handling Notes
- Maintain protein reagents at 4 °C
- Avoid prolonged incubation of streptavidin–biotin complexes before use
- Handle beads gently to prevent loss of immobilized synthetic cells
- Keep liposome samples at 30 °C only during active incubation steps
Immobilization of Synthetic Cells on Magnetic Beads (DBCO–Azide Coupling)
- Purpose
This protocol describes covalent immobilization of azide-functionalized synthetic cell liposomes onto DBCO-functionalized magnetic beads via click chemistry, including bead blocking to prevent post-division re-binding of daughter vesicles.
- Materials
2.1 Lipids and liposomes
- Synthetic cells prepared as in standard liposome protocol (Methods section 4)
- 16:0 azidocaproyl PE (Avanti 870126P)
- 1 mol% incorporated into liposome membranes
2.2 Magnetic beads
- DBCO-tagged magnetic beads (Kerafast FCC433)
- Binding capacity:
- 30–50 nmol DBCO per mg beads
2.3 Blocking reagent
- 3-Azido-L-alanine (Jena CLK-AA003)
- Prepared in 50 mM HEPES pH 8.0 (50 mM stock)
2.4 Buffers and lysis reagents
- 50 mM HEPES, pH 8.0
- Triton X-100 (0.1%; Acros Organics AC215682500)
2.5 Molecular biology reagents
- qPCR reagents targeting αHL plasmid
2.6 Equipment
- Magnetic rack / PCR tube magnetic stand
- Incubator at 30 °C
- Shaker for gentle mixing
- Lyophilizer
- qPCR system
- Preparation of Azide-Functionalized Liposomes
- Prepare liposomes using standard protocol (Methods section 4).
- Include:
- 1 mol% 16:0 azidocaproyl PE
- Stock preparation:
- 0.1 mg/mL azido lipid stock in chloroform
- Add:
- 2.54 µL per 30 µL sample (1 mM liposomes) during thin film formation
- Final azide concentration:
- Bead Preparation
- Wash DBCO beads:
- 3× with 50 mM HEPES pH 8.0
- Estimate functional groups:
- ~250 nmol DBCO in 5 mg beads
- Liposome–Bead Coupling Reaction
- Combine per reaction:
- 5 mg DBCO magnetic beads
- 30 µL azide-labeled liposomes
- Reaction stoichiometry:
- Liposomes: ~0.3 nmol azide total
- Note: ~50% accessible due to bilayer leaflet distribution
- Large excess of DBCO ensures complete capture
- Incubation:
- 30 °C for 8 hours
- Gentle shaking
- During incubation:
- Synthetic cell internal reactions (e.g., translation) may be initiated simultaneously if required
- Immobilization Verification (Control Assay)
- Magnetically separate beads
- Collect:
- Supernatant (“free fraction”)
- Lyse remaining bead-bound liposomes:
- Add 20 µL:
- 50 mM HEPES pH 8.0
- 0.1% Triton X-100
- Incubate to ensure complete lysis
- Process both fractions:
- Analyze via qPCR:
- Expected result:
- Bead fraction: full recovery of αHL plasmid (≈10 nM equivalent input)
- Supernatant: no detectable αHL signal
- DBCO Blocking to Prevent Rebinding of Daughter Cells
7.1 Rationale
Excess DBCO groups remain on beads after immobilization and may capture newly formed daughter liposomes, interfering with division experiments.
7.2 Blocking reaction
- After 8-hour immobilization step, add:
- 7 µL of 50 mM 3-Azido-L-alanine solution
- Final azide added: 350 nmol
- Incubation:
- 4 hours
- 30 °C
- Gentle shaking
- Outcome:
- Unreacted DBCO sites are quenched via azide coupling
- Post-Blocking Usage
- After blocking, bead–liposome complexes are considered ready for:
- Division experiments
- Growth and feeder liposome assays
- Competitive population studies
- Critical Handling Notes
- Maintain excess DBCO-to-azide ratio for full immobilization
- Ensure complete bead washing prior to coupling
- Avoid bead aggregation during incubation
- Always perform blocking step before division experiments
- Handle azide compounds carefully due to reactivity