What is SpudCell
SpudCell is a liposome encapsulated synthetic cell that can grow and replicate.
It undergoes a full cell cycle, with DNA replication, genetically encoded feeding and genetically encoded division.
What is SpudCell
SpudCell is a liposome encapsulated synthetic cell that can grow and replicate.
It undergoes a full cell cycle, with DNA replication, genetically encoded feeding and genetically encoded division.
Protocols for SpudCell formation and experiments
Below is the list of protocols and procedures needed to assemble and analyze a SpudCell.
Read this first
Synthetic cell experiments are still more resource-intensive than growing natural cells. While our motivation for this research is to make biology a general purpose technology, usable freely by all, we are currently operating in the sandbox environment.
To grow a SpudCell, your lab needs to have core competencies in:
If you haven’t used any of those techniques before, we strongly suggest you successfully set up those first, before moving on to SpudCell experiments.
pREP Transcription–Translation and Phi29 Replication Reaction
This protocol describes the preparation and incubation of the pREP cell-free transcription–translation and DNA replication system used for protein expression, Phi29 genome replication, and related synthetic cell experiments.
2.1 DNA preparation
2.2 Commercial reagents
3.1 10× Energy Mix
Prepare and aliquot:
3.2 50× rNTP Mix
Prepare and aliquot:
4.1 Final reaction composition (per 100 µL reaction)
Prepare reactions on ice.
|
Component |
Final concentration |
Volume (per 100 µL) |
|
10× Energy Mix |
1× |
10 µL |
|
50× rNTP Mix |
1× |
2 µL |
|
PURExpress Solution A |
0.1× |
2 µL |
|
PURExpress Solution B |
60% v/v |
60 µL |
|
dATP |
0.6 mM |
variable (from stock) |
|
dGTP |
0.6 mM |
variable |
|
dCTP |
0.6 mM |
variable |
|
dTTP |
0.6 mM |
variable |
|
DTT |
4 mM |
variable |
|
Phi29 DNA polymerase |
0.4 U/µL |
variable |
|
Plasmid DNA |
as required |
variable |
|
Nuclease-free water |
— |
to 100 µL |
5.1 Prepare bulk reaction master mix (excluding DNA and Phi29 polymerase if variable)
For N reactions, prepare:
5.2 Final assembly per reaction
For each 100 µL reaction:
Plasmid Preparation, Cloning, and Usage for pREP / In Vitro Translation Experiments
This protocol describes plasmid sourcing, cloning strategies, purification workflow, and concentration handling for DNA templates used in pREP transcription–translation and related in vitro expression systems.
2.1 External plasmid sources (non-lab-cloned constructs)
2.2 Promoter system standardization
3.1 General cloning methods used
All cloning was performed using one of the following:
4.1 Bacterial host strain
4.2 Workflow
5.1 Primary purification
5.2 Secondary purification
5.3 Rationale
6.1 Standard reaction concentrations
6.2 Multi-plasmid conditions
7.1 Problem addressed
7.2 Solution for high-plasmid load (>3 plasmids)
7.3 Outcome
8.1 DNA input integration
8.2 Mixing strategy
Synthetic Cell Liposome Preparation (Oil Extrusion Method)
This protocol describes the preparation, purification, and handling of synthetic cell liposomes used in cell-free synthetic biology experiments. Liposomes are prepared using a lipid thin-film method followed by oil hydration, emulsification, and differential centrifugation purification.
2.1 Lipids
2.2 Solutions
2.3 Equipment
3.1 Standard formulation (“1× liposomes”)
3.2 Final composition per 1 mM lipid sample
4.1 Preparation of stock solutions
4.2 Thin film formation (typical 30 µL liposome batch)
5.1 Lipid hydration in oil phase
6.1 Emulsion formation
7.1 First centrifugation (phase separation)
7.2 Liposome recovery and wash
7.3 Final cleanup
Liposome size is defined by extrusion membrane pore size during initial lipid processing:
Fluorescent Labeling of Synthetic Cell Liposomes
This protocol describes incorporation of fluorescent membrane dyes into lipid thin films for preparation of labeled synthetic cell liposomes used in imaging and tracking experiments.
2.1 Lipid dyes (chloroform stocks)
All dyes are used as 100 µg/mL stocks in chloroform:
3.1 Standard dye incorporation
3.2 Total dye loading rules
3.3 Effective concentration example
4.1 General procedure
For a standard 30 µL, 1 mM lipid preparation, add the following dye stock volumes directly into the chloroform lipid mixture:
|
Dye |
Stock concentration |
Volume per thin film |
|
NBD-PE |
100 µg/mL (chloroform) |
2.86 µL |
|
Rhodamine-PE |
100 µg/mL (chloroform) |
4.0 µL |
|
Cy5-PE |
100 µg/mL (chloroform) |
3.79 µL |
|
Cy7-PE |
100 µg/mL (chloroform) |
3.98 µL |
After dye incorporation, liposomes are prepared using the standard oil extrusion protocol:
Preparation of Feeder Liposomes for Synthetic Cell Growth and Division Experiments
This protocol describes the preparation of Ni-NTA–functionalized feeder liposomes used in synthetic cell growth, division, and generation-tracking experiments. Feeder liposomes are designed to deliver biochemical components and enable membrane-mediated interactions such as fusion and cargo exchange.
2.1 Lipids
2.2 Lipid stock solutions
2.3 Equipment
3.1 Base composition (all feeder liposomes)
3.2 Functional lipid addition
3.3 Example calculation
4.1 Lipid mixture preparation
4.2 Example (30 µL, 2 mM feeder liposomes, 0.6 mol% Ni-NTA)
4.3 Thin film formation
5.1 Standard formation
5.2 Hydration and emulsification
6.1 Purpose
6.2 Procedure
Each feeder liposome population may include:
Liposome Fusion Experiments
This protocol describes the preparation, tagging, mixing, incubation, and analysis of synthetic cell liposome fusion experiments using functionalized liposomes (e.g., protein tags, Ni-NTA systems, and αHL-mediated fusion).
2.1 Liposomes
2.2 Fusion tags and reagents
2.3 Chemicals
2.4 Equipment
3.1 Base preparation
3.2 Standard concentration
4.1 External fusion tag decoration
4.2 αHL-based fusion system
5.1 Standard mixing procedure
5.2 Mixing format
6.1 Standard conditions
7.1 Fluorescence measurement
7.2 Molecular analysis (if required)
FRET-Based Membrane Growth and Lipid Mixing Assay
This protocol describes fluorescence resonance energy transfer (FRET)–based quantification of membrane growth and lipid mixing in synthetic cell liposomes. The assay monitors dilution of membrane dyes upon liposome fusion and membrane expansion.
Membrane growth is quantified by measuring dilution of FRET pairs embedded in lipid bilayers. Upon fusion with unlabeled membranes, FRET efficiency decreases, leading to increased donor fluorescence.
3.1 Lipid dyes (FRET pairs)
3.2 Liposomes
3.3 Equipment
4.1 Pair usage
5.1 Liposome populations
5.2 Mixing principle
7.1 Purpose
7.2 Calibration series
Prepare control liposomes at defined dye dilution levels:
7.3 Preparation method
8.1 Signal acquisition
8.2 Spectral configuration
9.1 Signal readout
9.2 Calibration
Synthetic Cell Cycle with Feeder Liposome Growth and Mechanical Division
This protocol describes a multi-generation synthetic cell cycle system combining feeder-liposome–driven growth, pREP-based cell-free expression, and mechanical division via extrusion. The system supports tracking over 5 sequential generations with molecular and fluorescence-based readouts.
2.1 Liposomes
2.2 Reaction components
2.3 Reagents for analysis
2.4 Equipment
3.1 Liposome formation
3.2 Initial reaction composition (per 30 µL)
3.3 Loading conditions
5.1 First growth step (Generation 1 initiation)
Each feeder liposome batch contains:
7.1 End of each generation cycle
For each subsequent generation:
After each cycle (post-incubation + extrusion):
9.1 Fluorescence measurement
9.2 Liposome lysis for molecular assays
9.3 DNA digestion (new DNA quantification)
9.4 Sample concentration and preparation
11.1 Fluorescent protein control
11.2 No-GFP control condition
11.3 Outcome comparison
Generation Counter System (Oligo Ligation and Readout)
This protocol describes the assembly, optimization, and quantification of a synthetic “generation counter” system based on ligation of RNA oligonucleotides with complementary sticky ends. The system is used to track synthetic cell generations in liposome-based cell cycle experiments.
2.1 Oligonucleotides
2.2 Enzymes
2.3 Nucleotides
2.4 Buffer components (pREP-compatible ligation conditions)
2.5 Equipment
3.1 Stock preparation
3.2 Working mixture preparation
4.1 Standard reaction composition
Each 20 µL ligation reaction contains:
7.1 RT-qPCR quantification
7.2 Output measured
8.1 Synthetic cell (top strand loading)
8.2 Feeder liposome loading (bottom strand delivery)
Genetically Encoded Growth and Division (FLAG-tag–Mediated System)
This protocol describes a genetically encoded synthetic cell growth and division system using FLAG-tagged αHL, antibody–streptavidin linker chemistry, azide-functionalized membrane immobilization, and feeder liposome–driven growth.
2.1 Synthetic cells and liposomes
2.2 Genetic and protein components
2.3 Immobilization chemistry
2.4 Buffers and reagents
2.5 Solid support
2.6 Equipment
3.1 Biotin–FLAG antibody stock
3.2 Streptavidin working stock
5.1 Liposome composition
5.2 Immobilization setup
9.1 Post-incubation separation
10.1 “Free (off-beads)” fraction
10.2 “Bead-attached” fraction
Western Blot Analysis of Protein Expression
This protocol describes SDS-PAGE and Western blot analysis of proteins expressed in solution or within synthetic cell liposomes, including sample preparation, electrophoresis, transfer, immunodetection, and chemiluminescent imaging.
2.1 Samples
2.2 Lysis reagent (for liposome samples)
2.3 SDS-PAGE reagents
2.4 Transfer system
2.5 Immunoblotting reagents
2.6 Detection system
2.7 Equipment
3.1 Solution samples (no vesicles)
3.2 Liposome samples
Size Exclusion Chromatography (SEC) Purification of Liposomes
This protocol describes gravity-driven size exclusion chromatography (SEC) used to separate liposomes from unencapsulated solutes following synthetic cell or liposome preparation.
2.1 Chromatography column
2.2 Size exclusion resin
2.3 Buffers
2.4 Equipment
Single-Cell Plasmid and Generation Counter Product Abundance Analysis
This protocol describes isolation, dilution, and molecular screening of single synthetic cells to quantify plasmid distribution and generation counter products after growth and division cycles.
2.1 Synthetic cells
2.2 Molecular biology reagents
2.3 Equipment
3.1 Removal of feeder liposomes
3.2 Normalization
3.3 Single-cell dilution strategy
4.1 Rationale
4.2 Screening marker
For each putative single-cell aliquot:
8.1 Cell-containing selection
8.2 Quantification targets
Genetically Encoded Division of Synthetic Cells (Streptavidin–Biotin NTA System)
This protocol describes a genetically encoded division system for synthetic cells using streptavidin–biotin–NTA crosslinking to induce membrane organization and division-like behavior. The system supports both free-solution and immobilized bead-based formats, including competitive growth experiments.
2.1 Proteins and linkers
2.2 Buffers
2.3 Liposomes
2.4 Solid support
2.5 Molecular biology reagents
2.6 DNA markers (competitive experiments only)
2.7 Equipment
3.1 Streptavidin working stock
3.2 Biotin-NTA linker working stock
PART A — Free-Solution Division Experiments
7A. Reaction Setup
8A. Incubation
9A. Post-Incubation Processing
PART B — Immobilized Division Experiments
7B. Bead Immobilization Setup
8B. Incubation
9B. Fraction Separation
10B. Fraction Processing
10B.1 Daughter fraction
10B.2 Bead-bound fraction
PART C — Competitive Growth Experiments
12C. System Setup
13C. Genetic Markers
To distinguish populations:
Note:
14C. Competition Reaction
15C. Post-Reaction Processing
Immobilization of Synthetic Cells on Magnetic Beads (DBCO–Azide Coupling)
This protocol describes covalent immobilization of azide-functionalized synthetic cell liposomes onto DBCO-functionalized magnetic beads via click chemistry, including bead blocking to prevent post-division re-binding of daughter vesicles.
2.1 Lipids and liposomes
2.2 Magnetic beads
2.3 Blocking reagent
2.4 Buffers and lysis reagents
2.5 Molecular biology reagents
2.6 Equipment
7.1 Rationale
Excess DBCO groups remain on beads after immobilization and may capture newly formed daughter liposomes, interfering with division experiments.
7.2 Blocking reaction
This work was done in collaboration with the Engelhart Lab.
What is SpudCell
SpudCell is a liposome encapsulated synthetic cell that can grow and replicate.
It undergoes a full cell cycle, with DNA replication, genetically encoded feeding and genetically encoded division.
SpudCell page on Biotic website
Protocols for SpudCell formation and experiments
Below is the list of protocols and procedures needed to assemble and analyze a SpudCell.
Read this first
Synthetic cell experiments are still more resource-intensive than growing natural cells. While our motivation for this research is to make biology a general purpose technology, usable freely by all, we are currently operating in the sandbox environment.
To grow a SpudCell, your lab needs to have core competencies in:
If you haven’t used any of those techniques before, we strongly suggest you successfully set up those first, before moving on to SpudCell experiments.
pREP Transcription–Translation and Phi29 Replication Reaction
This protocol describes the preparation and incubation of the pREP cell-free transcription–translation and DNA replication system used for protein expression, Phi29 genome replication, and related synthetic cell experiments.
2.1 DNA preparation
2.2 Commercial reagents
3.1 10× Energy Mix
Prepare and aliquot:
3.2 50× rNTP Mix
Prepare and aliquot:
4.1 Final reaction composition (per 100 µL reaction)
Prepare reactions on ice.
|
Component |
Final concentration |
Volume (per 100 µL) |
|
10× Energy Mix |
1× |
10 µL |
|
50× rNTP Mix |
1× |
2 µL |
|
PURExpress Solution A |
0.1× |
2 µL |
|
PURExpress Solution B |
60% v/v |
60 µL |
|
dATP |
0.6 mM |
variable (from stock) |
|
dGTP |
0.6 mM |
variable |
|
dCTP |
0.6 mM |
variable |
|
dTTP |
0.6 mM |
variable |
|
DTT |
4 mM |
variable |
|
Phi29 DNA polymerase |
0.4 U/µL |
variable |
|
Plasmid DNA |
as required |
variable |
|
Nuclease-free water |
— |
to 100 µL |
5.1 Prepare bulk reaction master mix (excluding DNA and Phi29 polymerase if variable)
For N reactions, prepare:
5.2 Final assembly per reaction
For each 100 µL reaction:
Plasmid Preparation, Cloning, and Usage for pREP / In Vitro Translation Experiments
This protocol describes plasmid sourcing, cloning strategies, purification workflow, and concentration handling for DNA templates used in pREP transcription–translation and related in vitro expression systems.
2.1 External plasmid sources (non-lab-cloned constructs)
2.2 Promoter system standardization
3.1 General cloning methods used
All cloning was performed using one of the following:
4.1 Bacterial host strain
4.2 Workflow
5.1 Primary purification
5.2 Secondary purification
5.3 Rationale
6.1 Standard reaction concentrations
6.2 Multi-plasmid conditions
7.1 Problem addressed
7.2 Solution for high-plasmid load (>3 plasmids)
7.3 Outcome
8.1 DNA input integration
8.2 Mixing strategy
Synthetic Cell Liposome Preparation (Oil Extrusion Method)
This protocol describes the preparation, purification, and handling of synthetic cell liposomes used in cell-free synthetic biology experiments. Liposomes are prepared using a lipid thin-film method followed by oil hydration, emulsification, and differential centrifugation purification.
2.1 Lipids
2.2 Solutions
2.3 Equipment
3.1 Standard formulation (“1× liposomes”)
3.2 Final composition per 1 mM lipid sample
4.1 Preparation of stock solutions
4.2 Thin film formation (typical 30 µL liposome batch)
5.1 Lipid hydration in oil phase
6.1 Emulsion formation
7.1 First centrifugation (phase separation)
7.2 Liposome recovery and wash
7.3 Final cleanup
Liposome size is defined by extrusion membrane pore size during initial lipid processing:
Fluorescent Labeling of Synthetic Cell Liposomes
This protocol describes incorporation of fluorescent membrane dyes into lipid thin films for preparation of labeled synthetic cell liposomes used in imaging and tracking experiments.
2.1 Lipid dyes (chloroform stocks)
All dyes are used as 100 µg/mL stocks in chloroform:
3.1 Standard dye incorporation
3.2 Total dye loading rules
3.3 Effective concentration example
4.1 General procedure
For a standard 30 µL, 1 mM lipid preparation, add the following dye stock volumes directly into the chloroform lipid mixture:
|
Dye |
Stock concentration |
Volume per thin film |
|
NBD-PE |
100 µg/mL (chloroform) |
2.86 µL |
|
Rhodamine-PE |
100 µg/mL (chloroform) |
4.0 µL |
|
Cy5-PE |
100 µg/mL (chloroform) |
3.79 µL |
|
Cy7-PE |
100 µg/mL (chloroform) |
3.98 µL |
After dye incorporation, liposomes are prepared using the standard oil extrusion protocol:
Preparation of Feeder Liposomes for Synthetic Cell Growth and Division Experiments
This protocol describes the preparation of Ni-NTA–functionalized feeder liposomes used in synthetic cell growth, division, and generation-tracking experiments. Feeder liposomes are designed to deliver biochemical components and enable membrane-mediated interactions such as fusion and cargo exchange.
2.1 Lipids
2.2 Lipid stock solutions
2.3 Equipment
3.1 Base composition (all feeder liposomes)
3.2 Functional lipid addition
3.3 Example calculation
4.1 Lipid mixture preparation
4.2 Example (30 µL, 2 mM feeder liposomes, 0.6 mol% Ni-NTA)
4.3 Thin film formation
5.1 Standard formation
5.2 Hydration and emulsification
6.1 Purpose
6.2 Procedure
Each feeder liposome population may include:
Liposome Fusion Experiments
This protocol describes the preparation, tagging, mixing, incubation, and analysis of synthetic cell liposome fusion experiments using functionalized liposomes (e.g., protein tags, Ni-NTA systems, and αHL-mediated fusion).
2.1 Liposomes
2.2 Fusion tags and reagents
2.3 Chemicals
2.4 Equipment
3.1 Base preparation
3.2 Standard concentration
4.1 External fusion tag decoration
4.2 αHL-based fusion system
5.1 Standard mixing procedure
5.2 Mixing format
6.1 Standard conditions
7.1 Fluorescence measurement
7.2 Molecular analysis (if required)
FRET-Based Membrane Growth and Lipid Mixing Assay
This protocol describes fluorescence resonance energy transfer (FRET)–based quantification of membrane growth and lipid mixing in synthetic cell liposomes. The assay monitors dilution of membrane dyes upon liposome fusion and membrane expansion.
Membrane growth is quantified by measuring dilution of FRET pairs embedded in lipid bilayers. Upon fusion with unlabeled membranes, FRET efficiency decreases, leading to increased donor fluorescence.
3.1 Lipid dyes (FRET pairs)
3.2 Liposomes
3.3 Equipment
4.1 Pair usage
5.1 Liposome populations
5.2 Mixing principle
7.1 Purpose
7.2 Calibration series
Prepare control liposomes at defined dye dilution levels:
7.3 Preparation method
8.1 Signal acquisition
8.2 Spectral configuration
9.1 Signal readout
9.2 Calibration
Synthetic Cell Cycle with Feeder Liposome Growth and Mechanical Division
This protocol describes a multi-generation synthetic cell cycle system combining feeder-liposome–driven growth, pREP-based cell-free expression, and mechanical division via extrusion. The system supports tracking over 5 sequential generations with molecular and fluorescence-based readouts.
2.1 Liposomes
2.2 Reaction components
2.3 Reagents for analysis
2.4 Equipment
3.1 Liposome formation
3.2 Initial reaction composition (per 30 µL)
3.3 Loading conditions
5.1 First growth step (Generation 1 initiation)
Each feeder liposome batch contains:
7.1 End of each generation cycle
For each subsequent generation:
After each cycle (post-incubation + extrusion):
9.1 Fluorescence measurement
9.2 Liposome lysis for molecular assays
9.3 DNA digestion (new DNA quantification)
9.4 Sample concentration and preparation
11.1 Fluorescent protein control
11.2 No-GFP control condition
11.3 Outcome comparison
Generation Counter System (Oligo Ligation and Readout)
This protocol describes the assembly, optimization, and quantification of a synthetic “generation counter” system based on ligation of RNA oligonucleotides with complementary sticky ends. The system is used to track synthetic cell generations in liposome-based cell cycle experiments.
2.1 Oligonucleotides
2.2 Enzymes
2.3 Nucleotides
2.4 Buffer components (pREP-compatible ligation conditions)
2.5 Equipment
3.1 Stock preparation
3.2 Working mixture preparation
4.1 Standard reaction composition
Each 20 µL ligation reaction contains:
7.1 RT-qPCR quantification
7.2 Output measured
8.1 Synthetic cell (top strand loading)
8.2 Feeder liposome loading (bottom strand delivery)
Genetically Encoded Growth and Division (FLAG-tag–Mediated System)
This protocol describes a genetically encoded synthetic cell growth and division system using FLAG-tagged αHL, antibody–streptavidin linker chemistry, azide-functionalized membrane immobilization, and feeder liposome–driven growth.
2.1 Synthetic cells and liposomes
2.2 Genetic and protein components
2.3 Immobilization chemistry
2.4 Buffers and reagents
2.5 Solid support
2.6 Equipment
3.1 Biotin–FLAG antibody stock
3.2 Streptavidin working stock
5.1 Liposome composition
5.2 Immobilization setup
9.1 Post-incubation separation
10.1 “Free (off-beads)” fraction
10.2 “Bead-attached” fraction
Western Blot Analysis of Protein Expression
This protocol describes SDS-PAGE and Western blot analysis of proteins expressed in solution or within synthetic cell liposomes, including sample preparation, electrophoresis, transfer, immunodetection, and chemiluminescent imaging.
2.1 Samples
2.2 Lysis reagent (for liposome samples)
2.3 SDS-PAGE reagents
2.4 Transfer system
2.5 Immunoblotting reagents
2.6 Detection system
2.7 Equipment
3.1 Solution samples (no vesicles)
3.2 Liposome samples
Size Exclusion Chromatography (SEC) Purification of Liposomes
This protocol describes gravity-driven size exclusion chromatography (SEC) used to separate liposomes from unencapsulated solutes following synthetic cell or liposome preparation.
2.1 Chromatography column
2.2 Size exclusion resin
2.3 Buffers
2.4 Equipment
Single-Cell Plasmid and Generation Counter Product Abundance Analysis
This protocol describes isolation, dilution, and molecular screening of single synthetic cells to quantify plasmid distribution and generation counter products after growth and division cycles.
2.1 Synthetic cells
2.2 Molecular biology reagents
2.3 Equipment
3.1 Removal of feeder liposomes
3.2 Normalization
3.3 Single-cell dilution strategy
4.1 Rationale
4.2 Screening marker
For each putative single-cell aliquot:
8.1 Cell-containing selection
8.2 Quantification targets
Genetically Encoded Division of Synthetic Cells (Streptavidin–Biotin NTA System)
This protocol describes a genetically encoded division system for synthetic cells using streptavidin–biotin–NTA crosslinking to induce membrane organization and division-like behavior. The system supports both free-solution and immobilized bead-based formats, including competitive growth experiments.
2.1 Proteins and linkers
2.2 Buffers
2.3 Liposomes
2.4 Solid support
2.5 Molecular biology reagents
2.6 DNA markers (competitive experiments only)
2.7 Equipment
3.1 Streptavidin working stock
3.2 Biotin-NTA linker working stock
PART A — Free-Solution Division Experiments
7A. Reaction Setup
8A. Incubation
9A. Post-Incubation Processing
PART B — Immobilized Division Experiments
7B. Bead Immobilization Setup
8B. Incubation
9B. Fraction Separation
10B. Fraction Processing
10B.1 Daughter fraction
10B.2 Bead-bound fraction
PART C — Competitive Growth Experiments
12C. System Setup
13C. Genetic Markers
To distinguish populations:
Note:
14C. Competition Reaction
15C. Post-Reaction Processing
Immobilization of Synthetic Cells on Magnetic Beads (DBCO–Azide Coupling)
This protocol describes covalent immobilization of azide-functionalized synthetic cell liposomes onto DBCO-functionalized magnetic beads via click chemistry, including bead blocking to prevent post-division re-binding of daughter vesicles.
2.1 Lipids and liposomes
2.2 Magnetic beads
2.3 Blocking reagent
2.4 Buffers and lysis reagents
2.5 Molecular biology reagents
2.6 Equipment
7.1 Rationale
Excess DBCO groups remain on beads after immobilization and may capture newly formed daughter liposomes, interfering with division experiments.
7.2 Blocking reaction
TED talk on synthetic life
Kate presented concept of building synthetic minimal cells, with its biotechnological, biomedical and basic science implications, in a TEDx talk .
Kate’s article on personalised medicine using synthetic cell technologies
iBiology on synthetic cells:
Part 1: Synthetic Cells: Building Life to Understand It
Part 2: Synthetic Cells: Approach and Applications
We use synthetic cells to reconstitute and study natural biological processes, and to prototype and validate bioengineering tools that can be later used in live natural cells.
We developed and validated protein architecture which binds to single stranded RNA. Using this protein technology, we are developing tools for visualization and quantification of levels of expression of genes of interest. This tool will work by following the reconstitution of a protein probe upon interaction of sequence-specific RNA binding proteins with the mRNA of the gene of interest.
We are also aiming to edit the transcriptome of the gene of interest, selectively decreasing the level of expression of one splice variant (as opposed to cutting the DNA of the gene, which targets all splice variants indiscriminately).
This technology could potentially help in studying non-ER translation events, elucidating mechanisms of synaptic plasticity, as well as studying healthy and diseased translational profiles of genes, e.g., those involved in oncogenesis and other disease processes.
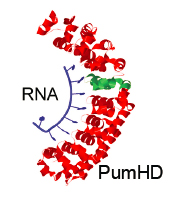
Programmable RNA-binding protein composed of repeats of a single modular unit;
Katarzyna P. Adamala*, Daniel A. Martin-Alarcon*, and Edward S. Boyden;
PNAS, 2016, 10.1073/pnas.1519368113; *equal contribution
local copy pdf
publisher website link

As we are approaching the limits of technological abilities of silicone microchips, biological computation is one of the alternatives for future development of computing devices.
Due to the complex and unpredictable nature of live cell biochemistry, we use simpler synthetic minimal cells to engineer and validate biological circuits.
![]()
We engineered biocomputing platform for constructing Boolean logic gates in synthetic cell system.
TRUMPET stands for Transcriptional RNA Universal Multi-Purpose GatE PlaTform.
The platform allows constructing orthogonal universal Boolean logic gates, including layering gates to engineer complex circuits. The logic gate design tool includes sequence validation and folding tests. More at trumpet.bio
We will build liposome based synthetic minimal cell based soft-computing circuits using genetic logic gates and sequential synthetic cell fusion. The proposed research will lead to a 105 scale-up of our ability to implement information processing in cells.
Large, complex genetic circuits will be implemented in both living cells and non-living vesicles, offering extremely dense and low-power computing. The proposed research spans the scale from computing in individual cells to that performed by multicellular ecological systems.
Here, insight is taken from software engineering to make genetics programmable. Key to our approach is refactoring genetics such that units of regulation are simple and modular and thus are able to be put together by EDA software.
In other words, it is not enough to build software to model genetics, rather it also requires building the genetics that can be put together by the software. This has led to new design principles in modularity and insulation, which we extend here by utilizing the principles of multicellular computing.
The use of non-living cell free systems and synthetic cells is a potential path towards systems that incorporate physical materials from biology and semiconductors.
 Origins of life
Origins of lifeThe earliest evolution of life included a series of transition from non-living matter, through prebiotic organic synthesis and chemical evolution, towards the Last Universal Common Ancestor of all life.
Our work focuses on the immediately-pre-life stage of evolution, when chemistry became biology.
We create synthetic minimal cells that exhibit some key properties of life, without being entirely alive. Those cells express proteins inside phospholipid liposomes, using cell-free protein expression systems. Thus, represent the latest stage of prebiotic evolution, after the establishment of the Central Dogma. Those cells do not exhibit active homeostasis, but they can maintain separate internal environment, they can grow, divide and evolve. The controllability and flexibility of those minimal cells allow us studying chemical processes underlying major transitions in evolution.
In our work, we create synthetic minimal cells expressing complex genetic pathways, with membrane proteins facilitating communication with external environment. Together, this creates a comprehensive system to study the advent of cellular processes on the boundary between prebiotic and Darwinian evolution.

Synthetic cells can be used as a chassis for biological and biomedical experiments under the conditions of space flight: performing reliable RNA and protein production and formation of drug particles will be crucial for establishing human presence on the Moon, Mars colony and for maintaining the health of people and animals during space flights and colonization. They can be prepared in advance and stored, lyophilised, for a long time.
We have developed synthetic cell epxerimenta payload, with a team of the RockSat-C 18 program. The payload was succesfully launched in a Terrier Orion sounding rocket from NASA Wallops in June 2018, increasing the TRL of this project to 6. This was, to our knowledge, the first time cell-free protein expression experiment was performed under spaceflight conditions.
Synthetic minimal cells have been proposed as a possible tool for biomanufacturing during long-term space missions and on extraterrestrial colonies, as well as a tool for future terraforming efforts. Synthetic minimal cells are bioreactors built specifically to mimic certain aspects of cellular processes, by reconstituting specific genetic pathways, encapsulated within a biomimic membrane. Thus, synthetic minimal cell technology combines most advantages of classical in vitro studies, with added capabilities, including spatial interactions, and interactions between many scaffolded and encapsulated proteins at once.
Traditionally, the information contained in the genetic material can travel only as far and as fast as the mass of the nucleic acids being duplicated during cell division. In the case of synthetic cells, since their autonomous reproduction is unnecessary, once a winning formula for a particular synthetic cell is discovered locally, it can simply be transmitted as digital information riding on optical fibers or radio waves at high speed (and using digital compression makes high bit rates and practically zero error rates feasible with currently available apparatus). Such information can be reduced back to synthetic cells anywhere with a synthetic cell generating apparatus ‐ be it in another country, spaceship or planet.
Our work focuses on developing mission architecture: preparation, storage and experimental conditions compatible with various flight scenarios. We are building syntetic cells to model various natural processes, to study elements of biology under the conditions of spaceflight. The primary goal of this research is to gather information about how natural terrestrial organisms behave under space flight conditions, for the benefit of designing human space exploration strategies. The other goal of this research is to help refine physicochemical boundaries for biochemistry as we know it, to aid in designing life detection systems.
Natural cells have a whole set of endogenous metabolic processes, pathways that use small molecules for suporrting the natural cell’s metabolism and growth. One of the biggest challenges in modern metabolis engineering is to integrate newly constructed pathways with the host metabolism. When trying to discover a new pathway, or improve efficiency of an existing enzyme pathway, we often run into the problem of not being able to tell whether the changes have the desired effect in the background noise of endogenous cell metabolism.
Synthetic minimal cells have no endogenous metabolism beyond what is engineered into those cells in the first place. Therefore, we can understand and control flux of small molecule substrates within the synthetic cell. This makes designing new pathways and optimizing existing ones much easier. Additionally, the short turnaround time of a synthetic cell experiment, without needing to wait for transformants to grow to test each new plasmid combination, make iterating metabolic engineering designs much faster.
Our research focuses on engineering methods for high throughput, combinatorial design of multi-gene pathways using synthetic cells as the chassis for testing and optimizing expression.
For large scale natural product synthesis, synthetic cells are mostly research and development tool, at this point it is still cheaper and easier to perform industrial scale production in a live bug. Synthetic cell screens can help identify candidate pathways, and optimize it before scaling up for industrial manufacturing.
Synthetic cell systems can be a good tool for small scale, pilot studies, or for rapid prototyping of on-demand production of small batches of biologicals or precious small molecule compounds.

During long term space missions, astronauts will encounter situations that are impossible to fully predict in advance. The isolation of the crew, for many years at a time, means that mission architecture needs to include plans for as many unpredictable situations as possible. The harsh environment of space and other planets will put high pressure on astronaut’s bodies. Providing on-demand, customizeable medical care under those conditions is very challenging.
We cannot plan for all pharmacy needs of astronauts by stocking the required drugs in advance. Long space missions require novel approach to producing pharmacticals on short notice from pre-defined, limited raw materials.
Synthetic minimal cells provide just the right kind of programmable bioreactors that can be used to manufacture small molecule and biological drugs in short amount of time. Synthetic cells can be lyophilised and stored for a long time, and the programmability of the synthetic cell genetic circuits means almost every kind of chemical transition can be coded with the appropriate enzyme, and new enzymatic pathways for manufacturing drugs can be created on demand.
Our research focuses on designing mission-oriented solutions for preparation and storage of synthetic cells for the purposes of small scale, fast and responsive manufacturing of pharmaceuticals.
Most of the efforts of biological research, both in studying healthy organisms as well as disease states, is focused either on studying the whole live cell (or organism), or on studying isolated reactions between few purified and well defined components in vitro. Most biological processes are not isolated events of interaction between few components, but rather complex interconnected networks built of often multifunctional nodes (proteins acting on many targets). The in vitro studies give results that are less relevant to natural biology – since an experiment with a few purified components does not acknowledge the vast complexity of a natural system. On the other hand, live cell studies are notoriously hard to reproduce and interpret, due to the variability between live subjects, as well as due to the inherent complexity of biology (cross-talk between the studied process and other pathways, or background signal from unrelated processes is often present).
Synthetic minimal cells deliver a solution bridging the existing gap between in vitro and live cell research: use synthetic minimal cells to investigate multicomponent gene pathways, combining the advantages of in vitro systems with the relevancy and complexity approaching that of whole cell studies.
Reading and controlling cells is the core purpose of modern synthetic biology, and the overarching goal of all biomedical studies. Both studying mechanisms of most diseases, as well as investigating healthy cellular processes, is currently done as either in vitro or live cell experiments. In vitro research methods are easy to use, cheap and efficient way to obtain information about behavior of specific, well defined protein or nucleic acid complexes or single enzymes, or to characterize small molecule interactions between metabolites or drugs and their specific biological targets. However, since life is structured in complexes that involve many components organized in precise 3D assemblies, the in vitro experiments often only deliver information about small snapshot of this complex, natural system. Studies of live cells allow to obtain truly biologically relevant information about complex pathways, but at significantly higher cost, and with results that are harder to interpret and often less reproducible. The variability between live cell subjects and the underlying intricacy of interconnected biological networks constantly interacting with each other makes signal measured in live cell experiments often more noisy and the experiment itself difficult to design.
Synthetic minimal cells offer a platform that allows studying complex genetic pathways, while keeping the complexity of the system at a level that still allows us understanding fully what the system contains and how to engineer it. Our research focuses on building tools for general use in many areas of synthetic biology, as well as studying some specific cases of complex biological processes, both healthy and diseased, that are not accessible by studying natural complex cells.
We are developing solutions for automating and scaling thropought of synthetic cell epxeriments. Microfluidic rigs help us work faster, iterating more genetic circuit designs with automated liposome formation and streamlined detection of protein expression.
Our first rig recently started producing droplets with fluorescent RNA.
We have developed a toolbox of computational resources, custom devices and protocols for engineering synthetic cells.
|
|
|
|
|
|
Addgene deposits from out lab (plasmids and bacteria strains):
www.addgene.org/Kate_Adamala/
Coming soon:
Cellulator
Tools are free to use; CC BY-NC-SA 4.0.
![]()
We have developed a toolbox of computational resources, custom devices and protocols for engineering synthetic cells.
|
|
|
|
|
|
Addgene deposits from out lab (plasmids and bacteria strains):
www.addgene.org/Kate_Adamala/
Coming soon:
Cellulator
Tools are free to use; CC BY-NC-SA 4.0.
![]()
This spreadsheet calculates number of liposomes, number of solute molecules per liposome, % of internal volume taken by the lumen of liposomes and volume per liposome.
You can also calculate number of molecules per liposome for embedding molecules in outer leaflet of the liposome.
To use this spreadsheet, you need to change minimum two values: lipid concentration and liposome diameter.
Please note the units! Lipid concentration is in millimoles (mM) and liposome diameter is in nanometers (nm).
Type the input values into the green fields.
Do not modify the blue fields unless you understand the equations and values used here.
If you make a mistake or want to start over, refresh the page.
You can download the spreadsheet using the download icon at the bottom of the frame.
This spreadsheet is calibrated for phospholipids only. Specifically, for POPC, with surface area of 0.7nm2. We used this spreadsheet for other phospholipids too, especially diacyl lipids with modified headgroups should have similar surface area.
If you want to do calculations for fatty acids, change the surface area (nm2) value to 0.35.
This spreadsheet makes the following assumptions: all liposomes are unilamelar, equal in size and spherical.
We would appreciate if you put our lab in acknowledgments when you use this spreadsheet for published work.
What is behind the spreadsheet
The efficiency of solute encapsulation inside POPC liposomes of a given radius r (nm) at a given concentration c (mM) can be estimated using this formula, used in the Szostak Laboratory and empirically confirmed by encapsulation experiments :
%internal volume = vol_liposome*liposomes_ml*10-19
Where:
vol_liposome = (4/3)*Pi*(r3)
is the volume of the lumen of a single liposome, in nm3;
liposomes_ml = surface_area_ml/area_liposome is the number of liposomes per 1 mL;
surface_area_ml = (c*10^-6)*((760*10^21)/0.9*NA)/2.5)/2
is the surface area of liposomes per 1 mL of solution of a given c (mM), with POPC MW=760 and length of the lipid bilayer approximated to 2.5nm; NA is Avogadro’s number;
and finally,
area_liposome = 4*Pi*(r2)
is the surface area of the liposome outer leaflet, in nm2.
These calculations were made with the assumption that liposome curvature is negligible, so the inner and outer leaflet contain an equal number of lipids and have equal surface area. The thickness of the bilayer was approximated at 2.5 nm. (1)
The addition of cholesterol increases bilayer thickness up to 30%, thus affecting the encapsulation rate (2) , but we cannot reliably estimate the influence of cholesterol on packing density and surface area of the liposomes.
According to this formula, a 25 mM solution of 200 nm POPC liposomes will contain ~14% of the total volume encapsulated inside liposomes.
In reality, the encapsulation rate of liposomes used in our experiments is likely lower. This is due to factors like the presence of cholesterol in POPC membranes and the fact, that in liposomes extruded through, e.g., a 200 nm filter, the size distribution of liposomes varies greatly and is, on average, smaller than 200nm. (3-5)
The differences in yield of protein synthesis inside synthetic cells, explained by the difference in efficiency of encapsulating the TX/TL enzyme mix, have been observed . (6)
1. Lewis, B. a & Engelman, D. M. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 166, 211–217 (1983).
2. Nezil, F. a. & Bloom, M. Combined influence of cholesterol and synthetic amphiphillic peptides upon bilayer thickness in model membranes. Biophys. J. 61, 1176–1183 (1992).
3. Jousma, H. et al. Characterization of liposomes. The influence of extrusion of multilamellar vesicles through polycarbonate membranes on particle size, particle size distribution and number of bilayers. Int. J. Pharm. 35, 263–274 (1987).
4. Olson, F., Hunt, C. a, Szoka, F. C., Vail, W. J. & Papahadjopoulos, D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta 557, 9–23 (1979).
5. Berger, N., Sachse, a., Bender, J., Schubert, R. & Brandl, M. Filter extrusion of liposomes using different devices: Comparison of liposome size, encapsulation efficiency, and process characteristics. Int. J. Pharm. 223, 55–68 (2001).
6. Caschera, F. & Noireaux, V. Compartmentalization of an all-E. coli Cell-Free Expression System for the Construction of a Minimal Cell. Artif. Life 22, 185-95 (2016).
The HTGAA course is based on the classic Fab-Lab “Hot to Make Almost Anything” fabrication class.
BioAcademy is the best resource to learn basisc of bioengineering, including specific applications of most cutting edge techniques. We run a synthetic cell module, including overwiew of the field of synthetic life and examples of synthetic cell based tools for biological engineering.

Build-a-Cell is an international community of scientists and policymakers working on building synthetic cells. We facilitate collaborations between groups in different areas of synthetic cell research, we engage with scientists in other disciplines to promote use of synthetic cell tools, and we provide information for the general public.
Our lab participated in every one of the bi-annual Build-a-Cell workshops, and we are actively pursuing collaborations with many Build-a-Cell members.
Kate Adamala is the co-founder and leader of Build-a-Cell.
STAT magazine published nice overview of efforts to build artificial life, From chemicals to life: Scientists try to build cells from scratch.
Details, including list of member labs, working groups, workshops and all resources can be found at www.buildacell.org.
The HTGAA course is based on the classic Fab-Lab “Hot to Make Almost Anything” fabrication class.
BioAcademy is the best resource to learn basisc of bioengineering, including specific applications of most cutting edge techniques. We run a synthetic cell module, including overwiew of the field of synthetic life and examples of synthetic cell based tools for biological engineering.
Excellent resource for beginning and advanced DIY biological engineers, biospaces and self-taught biohackers, as well as interesting supplement for anyone studying basics of modern biological engineering.
Astrobiology is studying past, present and future of the life on the Universe. Synthetic cells are the perfect tool to answer many of the questions typically asked by astrobiologists, including engineering synthetic cells to resemble earliest terrestrial life forms for studying of the origins of life on Earth, engineering synthetic cell systems to explore possibilities of biochemistries yielding life on other planets, and exploring possible future evolution scenarios for various life forms. Using synthetic cells we can ask many questions about “weird life” events and alternative life forms, questions that would be nearly impossible to answer studying modern, complex terrestrial cells.
Our work is driven by the question of what is the multidimensional niche space for life, and what are the molecular mechanisms of these boundary physicochemical conditions of life? This leads naturally to the questions of how does terrestrial life responds to changes at those boundary conditions, and can synthetic biology be used to expand these? We aim at defining comprehensive matrix describing mechanisms in which environment restricts basic processes of biology, and developing framework for studying and designing living systems around those limitations.

Understanding what separates life from non-life is critical to solving some of the great outstanding questions in science, such as how life first emerged and how we might unambiguously detect life on other worlds. The transition from non-living to living matter has been notoriously difficult to quantify, limiting our ability to develop the necessary theory for understanding life or its universal properties. One of the most significant challenges is the complexity of the examples of life available to study, which represent the product of more than 3.5 billion years of evolution. Systems representing the intermediate stages of complexity between simple chemistry and modern biology are only now becoming accessible to study in the lab with advances in systems chemistry and minimal synthetic life, respectively. These have not yet been connected to explore the uncharacterized landscape between simple chemical systems and the much more complex biochemical architectures characteristic of life today. Our project is an unprecedented opportunity to systematically study the full pathway from non-life to life through a synthesis of bottom-up chemistry, top-down minimal biology and fundamental theory. We aim to build very simple systems from chemical and biological parts, and by studying varying stages of programmed complexity and autonomy, systematically evaluate the transitions in information through which these become more life-like, self-referential entities.
We are developing solutions for automating and scaling thropought of synthetic cell epxeriments. Microfluidic rigs help us work faster, iterating more genetic circuit designs with automated liposome formation and streamlined detection of protein expression.
Our first rig recently started producing droplets with fluorescent RNA.